研究内容
特定の化合物から評価関数に基づき最適化する機械学習手法
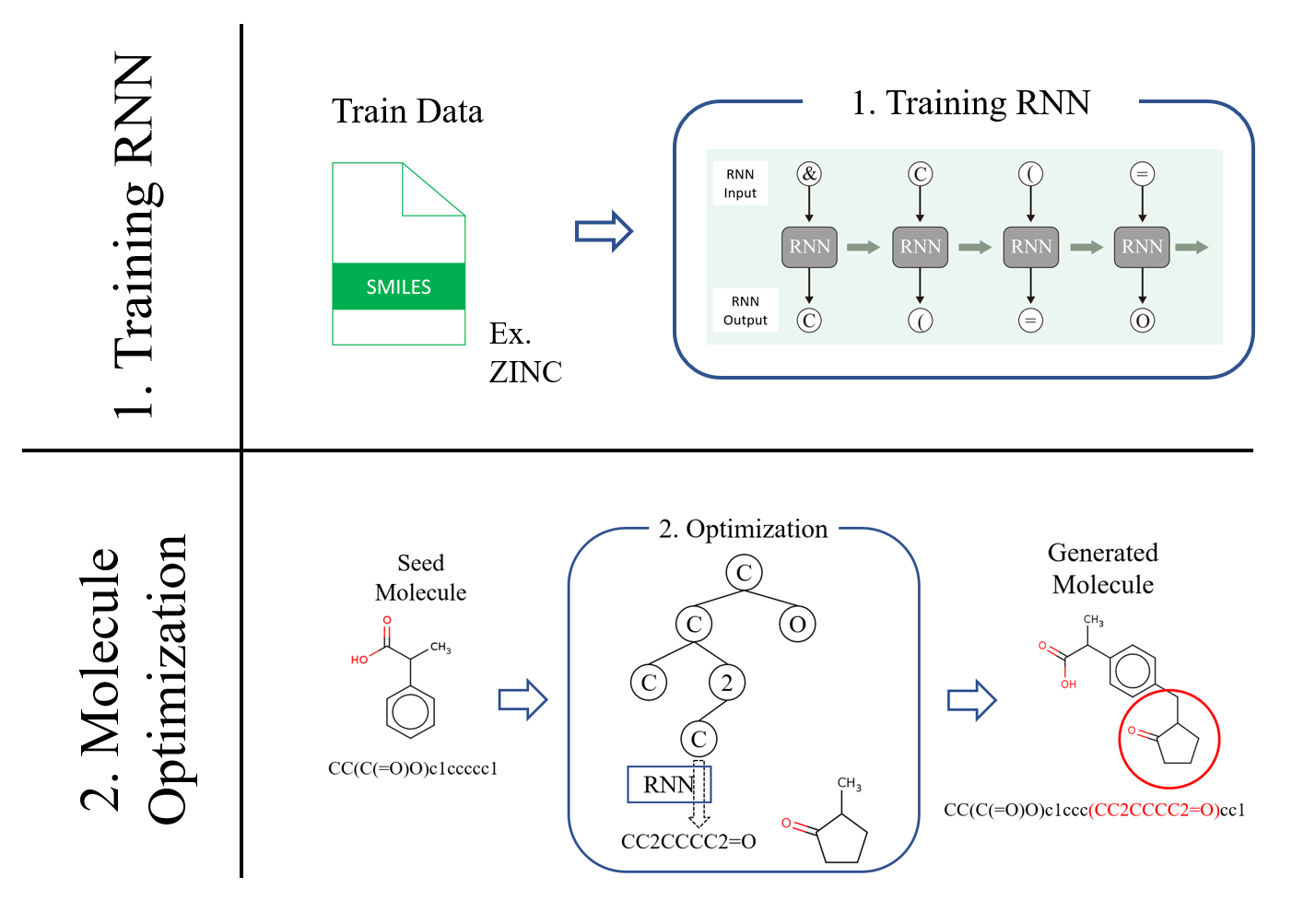
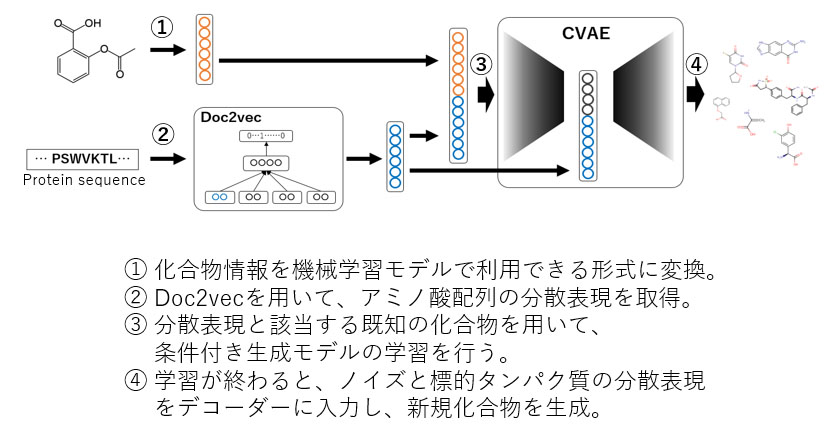
標的蛋白質のアミノ酸配列に応じた化合物生成モデル
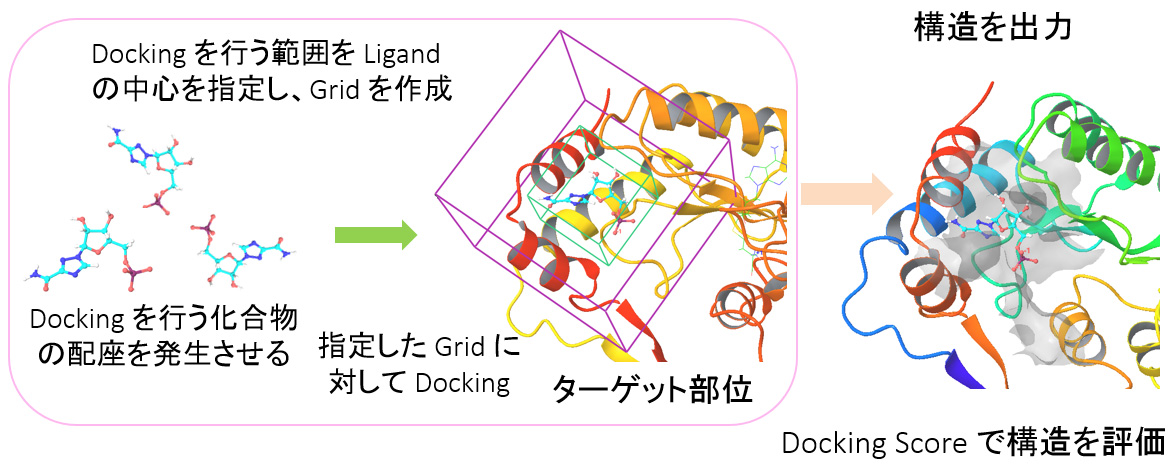
スマート創薬手法によるヒット化合物の発見
Mixed Reality(複合現実)による創薬支援システムの開発
 |
創薬において創薬標的タンパク質や薬候補化合物の立体構造の可視化は非常に重要となっています。しかし、現在多くの分子構造描画システムでは本来3次元の立体構造を2次元のディスプレイで描画しています。本来3次元であるタンパク質や薬候補化合物の立体構造は、その相互作用や化合物の最適化を考える上で3次元で可視化したほうが、より有用な知見が得られると考えています。そこで、本研究ではタンパク質や化合物の立体構造をMixed Reality(複合現実) を実現するデバイスであるHoloLens を用いて複合現実で描画するシステムHoloMolを開発しました。HoloMolはクラウドと連携することで、クラウドのデータをHoloMolで表示したり、クラウドを介して東京と筑波や、筑波とボストンというような遠隔でタンパク質や薬候補化合物の立体構造を共有することを可能にすることを目指しています。 |
CPU-GPU間の負荷分散に関する研究
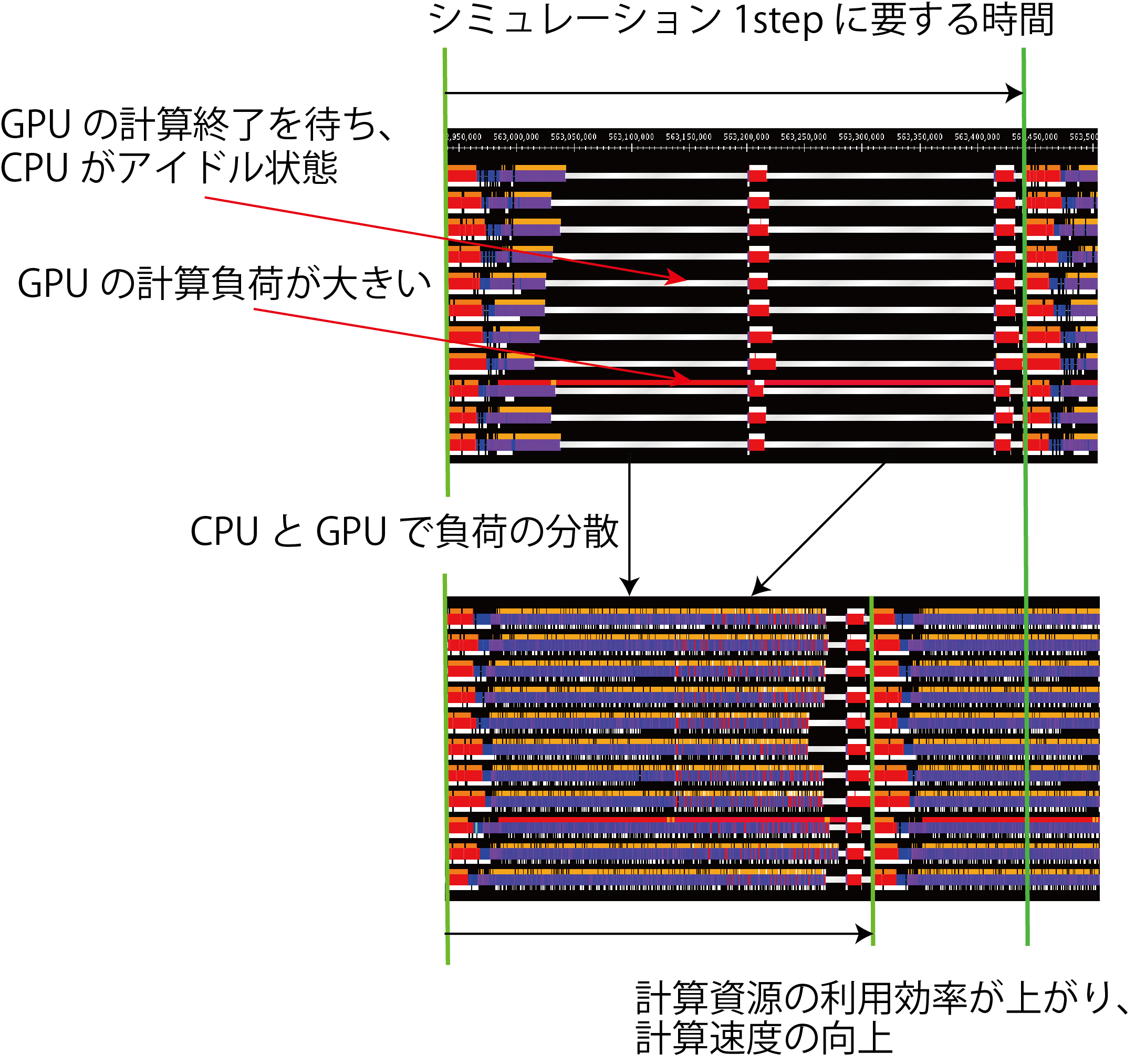 |
TSUBAME2.0や現在世界最速のスパコンTitanをはじめ、通常のCPUに加えてGPUなどのアクセラレータ、コプロセッサーを搭載する計算機が増えてきています。 こういった異なるプロセッサが混在する計算環境、すなわちヘテロジーニアスな計算環境においては計算資源を十分に活かすために様々な特殊な配慮が必要となります。その1つが異なるプロセッサ間で計算負荷のバランスをとることです。 図は分子動力学法のシミュレーションプログラムNAMDをTSUBAME上でCPUとGPUを用いて実行した際の、プロセッサの様子です。 |
参考:T. Udagawa and M. Sekijima, "GPU Accelerated Molecular Dynamics with Method of Heterogeneous Load Balancing," 2015 IEEE International Parallel and Distributed Processing Symposium Workshop, 2015, pp. 1008-1013, https://doi.org/10.1109/IPDPSW.2015.41.
タンパク質・リガンド間の結合自由エネルギー解析システムの開発
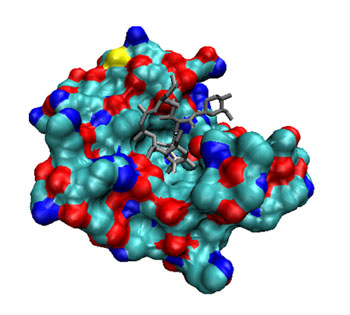 |
左の図は、タクロリムス(FK506)という免疫抑制剤の一種とFKBPというタンパク質の複合体です。FK506とFKBP複合体は、
カルシニューリン(CN)と結合することで、CN の脱リン酸化を抑制します。タンパク質は、他のタンパク質や化合物、ペプチドと結合することで様々な機能を発揮しますが、このようなタンパク質と結語する相手であるリガンドとの相互作用を求めることは非常に重要です。 本研究では、大規模計算機があって初めて可能になるタンパク質・リガンド間の結合自由エネルギーを解析するプログラムを構築し、創薬支援を行うことを目指しています。 ちなみに、FK506は筑波山の土壌細菌から見つけられたことでも日本では有名で、現在ではアトピー性皮膚炎の塗布薬にも使用されています。 |
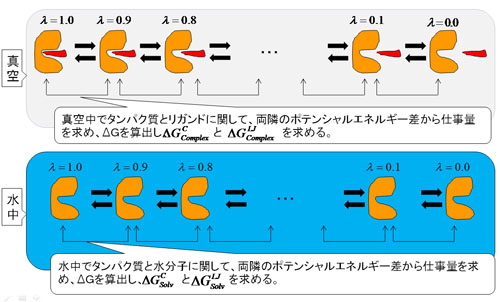
Van Der Waals力とCoulomb力の片方ずつに0〜1の間の変数λv、λcを掛け(掛けていない方の力のλは1で固定)、隣り合うλ間のポテンシャルエネルギー差から仕事量を求め、自由エネルギーを求めます。ポテンシャルエネルギーを求める際に、大規模な分子動力学シミュレーションを行います。W=U(λj, x)-(λi, x)により、ポテンシャルエネルギー差から仕事量を求め、Jarzynskiの式exp(-ΔG/kBT)=<exp(-W/kBT)>により、仕事量から結合自由エネルギーを求めることが出来ます。
確率的情報処理によるタンパク質の熱揺らぎ解析システムの開発
レプリカ交換法などのタンパク質の効率的な構造サンプリングを主眼におく方法では、自由エネルギー地形による解析は強力な手段になりますが、長時間一定の条件でフォールディング、アンフォールディングが観測できるシミュレーションが行えるなら、隠れマルコフモデルのような確率モデルによる解析が現実的になります。 現在、このような長時間のシミュレーションを行うことは多くの研究機関にとっては難しいことだとは思います。しかし、2011年に運用が開始される次世代スーパーコンピューターを始め、多くの研究者がいずれこのような大規模・長時間シミュレーション実行可能になる時が来ると考えています。その時に向けて、今まで行われていた自由エネルギー地形によるタンパク質のフォールディングや熱揺らぎの解析だけでなく、確率モデルによる解析手法等の今までのようなタンパク質のデータ一つ一つを見て議論する時代から、大量のシミュレーションデータから新しい価値を見いだす手法を開発したいと考えています。 また、実際に生化学実験を行っているグループと連携することで、本手法をタンパク質の設計や機能向上へも応用していきます。 |
|
 シミュレーションの初期構造 |
シミュレーションの条件 シニョリン(CM001) |
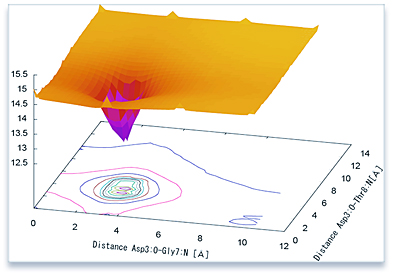 自由エネルギー地形による解析 |
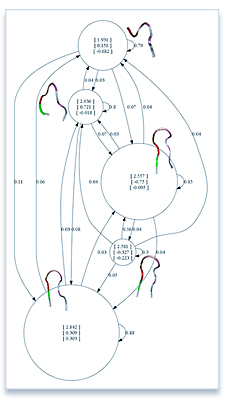 隠れマルコフモデルによる解析 |
正常な立体構造のプリオンタンパク質の異常構造への構造変化機構の解明
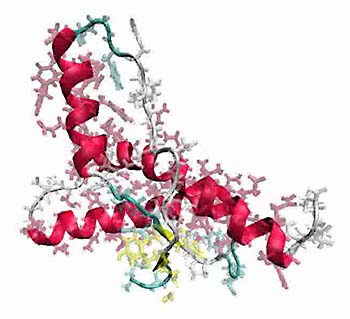 |
NMRで構造が決定されたヒトプリオンタンパク質 125-228残基 (プリオン病の発病には90-124残基も関連しますが、disorderである為に構造決定は為されていません。)上図をクリックするか、ココをクリックするとプリオンタンパク質の生体環境での振る舞いをアニメーション表示します。 |
プリオンタンパク質は、アミロイド化して難溶化することで狂牛病の原因となります。プリオンタンパク質は、アミノ酸配列が同じでありながら異なる立体構造をとるisoform(アイソフォーム)が存在していることが知られており、正常な立体構造をとっているものは生体内に元来存在していても、その機能は明らかになっていません。この正常な立体構造がmisfold(ミスフォールド)を起こして異常な構造のisoformとなることにより、アミロイド化・難溶化し、細胞内で沈着する。このことがいわゆるプリオン病と言われる狂牛病、スクレイピー、クロイツフェルト・ヤコブ病などを引き起こす原因となります
。しかしながら、この構造転移がどのようにして起き、プリオン病を引き起こすかは明らかになっていません。 |
|
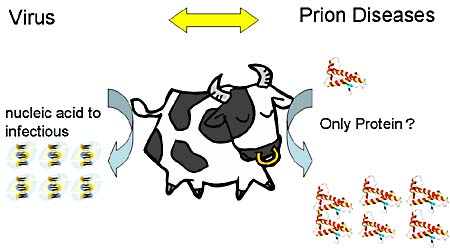 上図は、HIVやインフルエンザなどのウイルスとスクレイピーやクロイツフェルト・ヤコブ病などのプリオン病の感染の仕組みの違いを示す模式図です。ウイルスにおいては、感染に核酸が関与しています。しかし、プリオン病ではプリオンタンパク質だけ、もしくはプリオンタンパク質とファクターXと呼ばれる因子によってのみ感染が広がっていくと言われています。 |
|
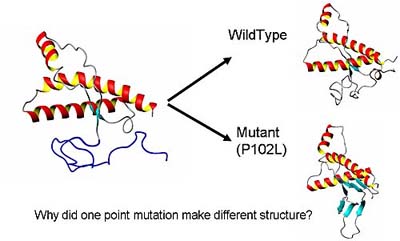 (左)初期構造、(右)シミュレーション後の構造 初期構造において異なっているのは102番目のアミノ酸残基がPro(野生型)かLeu(変異型)という点だけでも、このように僅か一残基の違いがプリオン病の発症に影響を及ぼすならば、その一残基がどのような影響を全体の構造に与えているか解析することは非常に重要だと考えています。このような研究は大規模なシミュレーションが出来て初めて可能になります。 |
|
design テンプレート